PART THREE
THE PROGNOSIS
Not everything that is new in health technology is good; not all that is good is needed.
Stephan Tanneberger "When Must a New Approach to Treatment Be Introduced?"
9
Managing the Medical Arms Race
In order to control the medical arms race, the policies that promote the flow of desirable innovations must be balanced with appropriate incentives that inhibit the production of unsafe technologies and the misuse or overuse of devices in the marketplace. Chapters 3 through 8 established the chronology of policy initiation and growth, outlining the evolution of public institutions and their interactions with the private sector. This chapter evaluates the consequences of policy proliferation. Recalling our analogy to polypharmacy, the discussion completes the brown-bag review.
This chapter is organized around the innovation continuum presented in chapter 1 (see figure 29). We can assume that desirable device innovations flow within the solid lines, all the way from the first idea to the patient's bedside. An appropriate policy mix would efficiently and expeditiously guide those products to the marketplace. Policies that restrict the flow, or allow them to spill over the banks, are unsuccessful.
The discussion in the chapter falls into three sections. The first provides an analysis of the present policy environment at both the discovery and the distribution stages, with illustrations of creative industry strategies that accommodate current policies. The second section discusses the pending and possible policy changes and their potential impact on the innovation stream. The third part addresses the interactions among the various public policies, which are inevitable consequences of policy proliferation. This part suggests mechanisms for eliminating
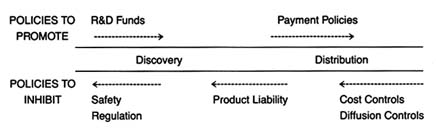
Figure 29. Policies affecting medical device innovation.
unnecessary duplication, reinforcing the effects of multiple policies, and encouraging coordination and cooperation among the relevant sources of policy, areas often overlooked by policymakers.
Where are We Now? Device Policy in 1990
Promoting Discovery
As discussed in chapter 3, the 1950s ushered in an era of belief in the social benefits of scientific and technological innovation. Biomedical research was seen as the foundation for improvements in health care quality—permitting new understanding of disease and innovative treatment for illness. The creation of the National Institutes of Health represented a major federal commitment to support of biomedical R&D.
It is difficult to measure the overall impact of this federal research policy on medical devices. The relevance of basic scientific research to subsequently developed products is often hard to trace. As we saw in chapter 3, however, some research may be clearly directed to specific technological or device development. Throughout the last forty years, there has been tension between those who advocate NIH funding for basic science exclusively and those who favor targeted programs. In the last twenty years, there has been an increase in directed or targeted programs with contracts let to medical device firms. The directed research at the NIH tends to fall into disease categories (for example, cancer, AIDS, and cardiac disease), rather than device categories
(for example, diagnostics or implants). For instance, NIH funding has supported innovation for treatment of renal diseases, which has led to artificial kidneys, dialyzers, and filtration systems, all of which are medical devices. However, not all supported research is disease-specific. Throughout the 1970s, NIH funded research on magnetic resonance imaging (MRI) systems, which can be used for diagnosis of many different medical conditions. The Small Business Innovation Research program also funds innovative private firms.[1]
The epigraph for part 3 is from Stephan Tanneberger, "When Must a New Approach to Treatment Be Introduced?" International Journal of Technology Assessment in Health Care 4 (1988), 113.
By 1982, however, industry was receiving only 6 percent of total NIH grants and contracts, and that sum included support for other activities in addition to medical device development.The industry has not been dependent on federal funds for R&D support. The private sector has played an important role in supporting research and development. Particularly since the late 1970s, there has been a growing number of venture capitalists, specialists in providing financial capital to small firms, who have promoted innovators. However, venture capital is particularly sensitive to economic trends and tends to shy away from supporting projects where development costs are high and pay-off is uncertain and longterm. Venture capital also considers the impact of future regulatory barriers—ease of entry encourages competition while premarket approval may be costly and time-consuming.[2]
Fred Dotzler and John Reher, "What Market Segments Attract Venture Capital Firms," Medical Device and Diagnostic Industry 11 (September 1989): 84-86.
Thus, NIH funds can serve as a bridge to support basic device research with the expectation that the private sector, through venture capital or other conventional fundraising efforts, will later oversee a project. In such situations, federal assistance can make or break a research project undertaken by a small firm. NIH refusal to fund can discourage research and development.[3]
See the discussion of the impact of NIH disapproval on cochlear implants in chapter 7.
Successful R&D Strategies: Novacor
NIH funding for the Artificial Heart Program illustrates the dramatic effects that government policy can have on particular firms. Novacor, a small innovative device company, owes its continued existence to NIH contracts. Dr. Peer Portnor, the founder of the company, is a physicist with experience in biomedical
research. Like other entrepreneurial biomedical scientists, he gravitated to research areas where government funding was available, thus illustrating the impact of targeted programs on the focus of R&D.[4]
Interview with Dr. Peer Portnor, December 1988.
The early work in his firm involved research in both the artificial heart, supported by the NIH, and gas analyzers, supported by NASA. Gradually, research came to focus primarily on the artificial heart, culminating in the 1978 formation of Novacor. Since that time the firm has received close to $20 million in NIH support. It has received some venture capital financing, but the long-term research agenda with limited commercial potential dampened the enthusiasm of early venture capitalists. Novacor was recently acquired by Baxter Travenol, but at least one-third of the firm's resources continue to come from the Artificial Heart Program.Venture capital showed early interest in Novacor, but NIH support has been consistent over the longer term. Novacor's left ventricle assist device (LVAD), or the Novacor Heart Assist System, provides a bridge to transplant for suitable candidates. It keeps patients with failing hearts alive until a transplant can be performed. The firm is also working on a fully implantable permanent heart replacement for patients who cannot tolerate a transplant. The system is still in experimental clinical research at major university hospitals.[5]
Spyros Andreopoulos, "A Helper for the Ailing Heart," Stanford Medicine (Fall 1986): 6-9.
The NIH is supporting a three-phase program for the fully implantable, electrically powered ventricle assist systems—device readiness, clinical evaluation, and patient follow-up. The first phase began in 1985. The AHP funded four groups of contractors, including Novacor, to run twelve LVAD systems for two years in a laboratory setting. At the second stage, the NIH planned to choose the two best preclinical performers for clinical trials. All three other firms experienced failures in the bench testing and dropped out of the trials. Two Novacor devices met the two-year performance testing requirement in December 1988. Consequently, Novacor was the only firm to receive a contract for the second clinical phase, with trials expected to begin in 1990 with fifty systems ready for implantations.[6]
Interview with Dr. Peer Portnor.
For firms that are successful, the benefits of NIH targeted device development are clear. The AHP broke some of the traditional
barriers to bioengineering that had developed at the NIH. But federal funds do not guarantee success. Many of the early innovators in the AHP did not survive despite federal support. Federal funds clearly can encourage certain avenues of research and can keep a research program alive when the private sector would have abandoned it.
In summary, there is widespread public acceptance of the federal government's efforts to support basic biomedical research. The direct federal policy promoting device discovery has been relatively modest. Device development is more likely to receive federal support if it can be used in the treatment of a disease that has been targeted by the NIH. The existence of NIH programs appears to spur and direct research in these areas as the story of Novacor indicates. The goal in many cases is to provide support for products not attractive to private capital because of long-term product development.
Inhibiting Discovery
The social motivation underlying federal regulation of medical devices was a desire for safer products. Following the controversies surrounding defective pacemakers and dangerous IUDs, it was clear that the public believed that the economic demands of the marketplace did not provide sufficient consideration of device safety. The prescription for this social ill was regulation, and the Medical Device Amendments of 1976 instituted an elaborate and comprehensive federal regulatory apparatus. Although the law covers all medical devices, its design intentionally focuses the highest level of scrutiny on a narrower group of high-risk devices.
Products requiring FDA premarket approval include devices for which sufficient data do not exist to ensure a reasonable degree of safety and efficacy. In practice, this requirement applies to less than 10 percent of all devices on the market. Devices subject to the requirement tend to be those that have serious consequences for patients if they fail. Examples include monitoring equipment and anesthesia delivery devices. Most implanted devices fall into the highest classification as well, including
heart valves, pacemakers, ocular lenses, and prostheses. Another device category subject to premarket approval includes products that are significant breakthrough technologies about which little has been known before the innovation. One example is lithotripsy, the ultrasonic kidney stone crushing device; others include magnetic resonance imaging (MRI) and CT scanners.
All these devices represent the kind of important innovations that can improve the quality of care. The public desire for safety led to the creation of market barriers for all innovations. Producers must spend time and money to comply with FDA requirements. For example, experts estimate that the 3M Company budgeted in excess of $15 million for safety and efficacy trials associated with the cochlear implant. Total costs, of course, vary depending upon the device.
Complete data on the impact of federal regulation, particularly premarket approval requirements, have never been collected. Some research indicates that regulation may deter smaller firms from undertaking innovation or force them to abandon the field. The case study on cochlear implants discussed in chapter 7 is just one example of the difficulties of compliance with regulation. For the vast majority of producers, however, compliance involves reporting and recordkeeping requirements that may be annoying but that do not involve the significant costs and delays associated with premarket approval applications.
The second major policy is product liability, which is also a reflection of the value of safety. Product liability offers retrospective compensation for harms associated with a product, with an expectation that the consequences of a lawsuit will deter the future production of unsafe products. It is very difficult to predict the impact of product liability prospectively because court decisions vary from state to state and are subject to constant modification through common-law evolution and statutory amendments.
If one had to predict the likelihood of liability problems, the following categories of devices would be relevant. While regulation looks to the device itself, product liability predictions depend on additional environmental or situational factors. Products
used by large numbers of patients have increased liability exposure; products used by certain classes of patients are more vulnerable than others. For example, young and otherwise healthy women injured by birth control devices invite sympathy from juries. Injuries to young children also generate large awards. Products used in high-risk procedures such as premature births, major surgery, or lifesaving situations are particularly vulnerable to litigation. The greater the risk to an otherwise healthy patient, the higher the likelihood that the technology will be implicated in an adverse court decision.
Regulation and product liability exposure target many of the same devices, but product liability has the additional consequence of proceeding case by case. Thus, one producer can face literally thousands of product liability suits with one product design failure. The case studies of pacemakers and the Dalkon Shield illustrate the devastating effects of mass lawsuits on the economic well-being of firms. However, even a small number of cases can be costly, and high expenses are incurred even when the producer prevails. Defending a successful case can run into tens of thousands or even hundreds of thousands of dollars. One punitive damage award can be several million dollars or more.
Creative Strategies: The IUD in the 1990s
The story of the IUD marketplace illustrates the effects of regulation and product liability on producers. As one might expect, producers will simply abandon the field if the environment is too threatening, as did several prominent IUD makers. Ortho Pharmaceutical withdrew its Lippes Loop in September 1985, and G. D. Searle halted U.S. sales of the Copper 7 IUD in January 1986. These two firms accounted for more than 95 percent of the IUD market at the time. Both cited business considerations, primarily lawsuits, not medical risks, as the reasons for withdrawal.[7]
The Population Council, news release, 28 October 1987.
Since its first IUD went on the market in 1974, Searle had won thirteen of fifteen IUD related lawsuits that reached the trial stage. The two adverse judgments totaled about $300,000. The cost of fighting four suits was over $1.5 million in 1985 alone.[8]Don Colburn, "Informed Consent Eases the Return of IUDs," Washington Post, 10 November 1987, 7.
Two smaller firms, however, recently developed creative responses to this less-than-optimal policy environment. Alza Corporation, a company that specializes in drug delivery systems, had offered an IUD that releases a birth control hormone. The IUD, known as Progestasert, entered the market in 1976. When Ortho and Searle left the field, Alza was the last firm selling IUDs in the U.S. market. It could have withdrawn as well. As a small company, it could ill afford major liability claims, and the Progestasert was not an especially important product in the firm's line. Alternatively, it could have aggressively marketed its product to seize market share.
Instead, Alza crafted a solution that would allow it to remain in the market while limiting its sales to women who would benefit from the product. The firm also consulted with women's health groups and legal advisers. The result is an unusually comprehensive program of physician and patient information. A patient counseling brochure requires potential patients to read and sign ten sections to indicate that they have discussed the material with the physicians and have read and understood the information. The physician or nurse is also required to sign a patient information form to indicate that counseling has taken place.
The firm has intentionally kept sales low—50,000 to 75,000 insertions a year. This number is significantly below the IUD market of over one million women in the mid-1970s. Responsible marketing has avoided sales to patients outside the recommended safety parameters, and detailed information helps to control potential liability suits brought on the basis of failure to warn of potential risks.[9]
"Progestasert: Intrauterine Progesterone Contraceptive System Patient Information" (Alza Corporation, 1986). Interview with Peter Carpenter, vice president of Alza Corporation, January 1987.
A second firm followed in Alza's footsteps at the end of the 1980s. Concerned about declining research on contraception in the litigious environment of the United States, the Population Council, a nonprofit research group, developed an IUD that received FDA approval in 1984. The product was originally to be sold by Searle, but that arrangement fell through when Searle left the IUD market.[10]
"Careful Patient Selection Allows for Return of Copper IUD to U.S. Market," American College of Obstetrics and Gynecology 32 (January 1988): 1, 11.
The Population Council then licensed the IUD technology to GynoPharma, a small start-up firm, in 1987. The firm submitted labeling and patient information to the FDAin 1988. ParaGard, the new product, is marketed with an extensive informed consent brochure modeled on Alza's document.
It is clear that regulation and product liability can have a powerful effect on the success or failure of medical devices. Products that are risky, even if they offer substantial benefits, must pass through significant FDA scrutiny. If they also have attributes that expose them to product liability (large patient base, high likelihood of harm to otherwise healthy individuals, for example), there can be additional potential barriers.
Balancing Promotion and Inhibition in Distribution
The present government policy toward distribution can promote or inhibit the flow of medical devices. As we saw in chapter 4, the advent of Medicare and Medicaid in the mid-1960s ushered in a period of expansion of health care services to large groups previously excluded from the system. The motivating social value behind these programs was a belief that there should be widespread access to the health care. The data indicate that medical technology sales soared following the advent of these government programs.
The policies of the 1970s and 1980s tried to contain or control costs associated with these federal and state programs. For some products, cost containment has not controlled market forces that encourage overdiffusion. When a policy works, however, cost containment protects the value of access by squeezing out unnecessary and wasteful expenditures incurred under the old cost-plus reimbursement system, expenditures that can lead to dangerous and unnecessary medical interventions. More cynical observers of HCFA's cost controls have argued that cost containment limits access and lowers the quality of care. The challenge has been to design a cost-efficient system that protects access without compromising quality. Results of the government's efforts to create a balance between the two have been mixed.
Thus, there was a new reality for medical device producers. There was the possibility of either a highly supportive or a very restrictive federal policy. When planning a new product introduction,
the industry has had to learn how to manage given extreme market uncertainty. As politicians and bureaucrats tinker with reimbursement rules, device company managers have had to adapt rapidly to the idiosyncrasies of the regulated marketplace.
The impact on medical device technologies varies. Relevant factors include the costs of acquiring the technology, the costs of using the technology, the alternative treatments available, the location in which the device is used, and the effect of the device on outcomes (an increasingly important factor given the growing trend toward outcomes measures).
As indicated in figure 30, the most significant negative impact has been on cost-raising technologies. Technologies that increase quality while lowering costs (upper left quadrant) tend to survive, while those products that raise costs without increasing quality or reducing quality (lower right quadrant) would have a harder time in the present policy environment. Technologies that increase both quality and cost, however, face a very uncertain future. Ideally, if the incremental costs are worth the incremental benefits, the product should succeed. The challenge is to design policies that accomplish these goals. Our present policy environment does not always do so.
Both providers and producers have pointed out that assessing the costs of a technology before introduction and diffusion is problematic. Some have argued that if traditional cost-effectiveness analysis has been applied, the CT scan would never have been approved. Ultimately, CTs have been a cost-saving diagnostic technology that has dramatically increased the quality of medical care. At the very least, firms must now gather data to justify the costs of their products and would be well served by presenting information on the impact of their products on health outcomes as well. The need for research on the effectiveness of new technologies remains high. Policymakers have made only modest commitments to technology assessment.
Creative Strategies for Cost Control
Some firms have inaugurated creative strategies in this environment, striving to raise quality while minimizing cost. One example
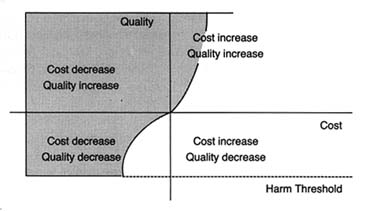
Figure 30.
Impact of cost containment on medical technology.
Shaded area represents positive effects."
Source: Nancy Cahill, esq.
is Acuson. Acuson designs, manufactures, and markets medical diagnostic ultrasound imaging equipment and has expanded sales throughout the 1980s. Its technology has several features that have helped it adapt to the cost-contained marketplace. The company's philosophy has been to develop products that are innovative, versatile, and capable of being upgraded or adapted to additional applications. For example, there are two major ultrasound imaging formats—sector and rectilinear. The former provides a wedge-shaped field of view, the latter a rectangular field. There are also three major ultrasound modes: imaging, Doppler (for analyzing blood flow), and M-mode (for cardiac analysis). Acuson's computed sonography system not only offers significantly better image quality but also provides films in both imaging formats and operates in all three modes. It can be upgraded primarily through software development without expensive investments in new hardware.
In 1985 and 1986, the firm added improvements without rendering its basic system obsolete or requiring customers to make major hardware changes. This flexibility allowed hospitals to acquire breakthrough technology efficiently. Acuson's system was identified as one of the products that "won big" in 1987.[11]
Gary Stephenson and Greg Freiherr, "Products that Won Big in 1987," Medical Device and Diagnostic Industry 10 (July 1988): 26, n. 13.
Thus, it has managed to compete with a number of domestic firms, including major players such as General Electric andHewlett-Packard, as well such important producers in the international marketplace as Hitachi Instruments, Philips Ultrasound, Siemens Medical Labs, and Toshiba Medical Systems. Many of these competitors have significantly greater financial and other resources and compete in more market segments than Acuson.
As the discussion makes clear, some products have attributes that minimize the complications and the barriers of the policy environment. Low-risk, cost-reducing products find a hospitable marketplace. However, many of our most desirable innovations are expensive to develop, may bring high risks along with their benefits, and may raise costs. The failure of the cochlear implant illustrates the sensitivity of the device industry to public policy. If the market is perceived by the innovators as too hostile, then technologies will fall by the wayside. Indeed, there is evidence in some fields, such as contraception, anesthesia, and vaccines, that innovation has been adversely affected.
On the other hand, there is also evidence of overdiffusion in the medical marketplace, at least for some technologies. Over-diffusion leads to misuse and abuse of medical technology, adding unnecessary risks and additional costs to the health care system. This phenomenon occurs because of the attributes of the medical market and the limited ability of public policy to control market failures.
Demand for medical technology is enormous and often undisciplined by the market. Providers of care are attracted to new technologies because of economic incentives, professional pride, and peer pressure to be innovators and leaders in their field. These professionals, in turn, are pressured by equipment manufacturers who tout the virtues of their products and, on occasion, by patients who hear about new treatments in the media.
These groups often overlook costs. Patients with third-party coverage are insensitive to price, and providers expect reimbursement for services they perform. Moreover, traditional referral networks among specialists are often based on patterns of trust among professionals without reference to comparative costs of services provided. Sometimes, too, fear of medical malpractice drives providers to order additional tests and procedures.
Of course, excess demand can be constrained by limited supply. As we have seen, efforts to control supply through certificate-of-need programs in the states have been relatively unsuccessful. Supply can also be controlled by third-party payers. If the payers refuse to cover a new procedure or technology, that treatment will effectively not be available to patients. Medicare and Medicaid are large payers that have an impact on market supply. As we saw in chapter 7, Medicare coverage policy is decentralized and fragmented. Regional carriers are subject to pressures from patients and providers demanding access to new procedures and technologies. Under pressure, these payers tend to cover the desired procedure.
Once covered, third-party reimbursement rates become crucial. As we have seen in the case of cochlear implants, a disadvantageous pricing decision can destroy a technology. Conversely, expectation of high profits often encourages overuse and misuse, as the case studies of the pacemakers and intraocular lenses illustrate.
Public payers are hampered in their coverage and payment decisions by inadequate information. Assessment of the costs and benefits of new technology is problematic. The need for more information on the effectiveness and costs of new technologies is high; the federal government has been unwilling to invest sufficient resources for assessment.
And, once profitability of a procedure is established, greed often follows need. Acquisition of equipment or special skills associated with a new technology or procedure create economic pressures. There is increasing evidence of supply-induced demand, particularly where demand can be manipulated by physicians and hospitals anxious to turn a profit on new equipment. Examples of excess supply include the proliferation of CT and MRI equipment and the overcapacity of mammography screening devices. The aggregate data support a conclusion of over-diffusion, since the United States alone consumes about one-half of the world's supply of medical technology.
This vicious cycle of demand fueling supply, then supply inducing demand is what constitutes the medical arms race. Public payers, such as HCFA, can affect this process, although its market share is too small to exert a strong influence. Congress
and HCFA have demonstrated neither a sufficient understanding of this process nor the political will to constrain providers and producers.
Where are We Going? Changing Prescriptions through Policy Reform
Reformers obviously believe that there are some problems in the policy environment and seek to correct them. To understand motives for policy reform, we need to evaluate all the essential components of the problems, from the initial symptoms to the present cures. The symptoms are the social problems perceived when behavior in the private sector conflicts with shared social values, such as a desire for safe products or quality health care. Public policies, or prescriptions, are the government's way of imposing those values on the private sector. The manner in which the policies are crafted and implemented leads to effects on the device industry. These effects may be further influenced by the interactions of various policy prescriptions.
Treatment options depend upon an interest group's evaluation of each step of the inquiry. If reform is proposed, is it because the effects of the policy were unanticipated? If an effect is undesirable, is it because of a flawed policy design or a failure of implementation? Reform proposals often call into question the underlying values that first generated the policy.
This section will describe the various policy reform proposals under consideration in 1990. Several important themes emerge. First, there appears to be nearly unanimous acceptance of the federal government in all the areas under discussion; no viable interest group advocates that it abdicate its various roles. Thus, government participation in all aspects of device innovation is now legitimate. Second, there is little coordination among the described proposals. The effect of policy interactions, an important concern for polypharmacy, is generally overlooked in the present policy environment. Moreover, different interest groups dominate different policy reform arenas, so that greater inconsistencies in policy may well arise over time.
Policy Reform at NIH
The NIH is fully accepted as a legitimate supporter of biomedical research. However, there is controversy about how much funding to allot to the NIH and how to allocate money among the many competing areas of research.
NIH funding requires congressional authorization. In addition, the National Cancer Institute and the National Heart, Lung, and Blood Institute require specific periodic reauthorization. Budget debates at the NIH are constant and highly political. At any given time, the NIH is concerned simultaneously with spending its current budget, steering proposals through the executive and legislative branches, and developing a budget for the next funding cycle.[12]
For an interesting British view of the NIH and its political environment, see Richard Smith, "Glimpses of the National Institutes of Health: Funding and Structure," British Medical Journal 296 (27 February 1988): 631-634. Specific programs often are introduced because of the personal interest of particular congressional members. In 1988, an amendment to create a new institute on deafness was offered by Tom Harkin, who has a deaf brother and is chair of the House Subcommittee on the Handicapped. Opponents argued against proliferation of institute-level components within the NIH.
The battles over budget size can be bloody. For example, the Reagan administration consistently tried to cut back congressional proposals for NIH increases. When the NCI and the NHLBI came up for reauthorization in 1985, President Reagan twice vetoed reauthorization measures, only to see his second veto overridden by huge margins (380-32 in the House and 89-7 in the Senate).[13]Ibid.
The medical device industry is generally supportive of large NIH budgets. During the 1989 Senate hearings on the NIH budget request, Frank Samuel, then president of HIMA, testified in support of the requested funding level of $8.2 billion. He acknowledged the debt of the industry to the NIH by listing innovative products that NIH funding supported. He specifically mentioned the Small Business Innovation Research Program (SBIR) as beneficial to the device industry.[14]
Senate Subcommittee on Labor, Health and Human Services, Education and Related Agencies, Committee on Appropriations. Statement of Frank E. Samuel, Jr., president, Health Industry Manufacturers Association, on the 1989 fiscal year budget request of the National Institutes of Health, 24 May 1988.
The device industry favors programs targeted to firms but has not challenged the basic research performed at the NIH.There is no compelling demand for major NIH reforms from any quarter: no general outcry for more money for medical device technology research or concern over the general types of research supported by the NIH. However, issues concerning American capacity for innovation have been voiced in relation to technology in general. It is possible, indeed likely, that innovation may be affected by cost containment in the public and private sectors, which will reduce corporate profits and R&D
expenditures. The recession of the early 1990s may also limit the ready availability of private venture capital funds, particularly if short-term profitability of new technology is limited. In order to enhance competitiveness and encourage innovation in such an environment, pressure may grow for greater government involvement.
This pressure could take several forms. First, there may be a push for more money for technology research within the existing NIH framework, such as more targeted funds for specific technologies not being aggressively developed in the private sector. Congress could create a new institute for medical technology development at the NIH that could coordinate and oversee the medical device research being performed there. While some oppose proliferation of institutes at the NIH and others see devices as simply part of the disease-oriented institute structure, more coherent technology research is needed at the federal level. This need, as we have seen, may deepen in an economic environment not conducive to innovation. How such an institute might interact with other federal institutions will be discussed later in this chapter.
Policy Reform at FDA
The value of safety underlies liability and regulation. The debate at the federal level about FDA regulatory reform has been dominated by members of Congress determined to tighten the regulatory rules applicable to devices and supported by proconsumer organizations such as Nader's Health Research Group. They assert that the FDA has failed to implement the 1976 law adequately and that the agency should administer a stronger dose to the industry. The 1990 Safe Medical Devices Act discussed below is the result of years of pressure to reform device regulation. In contrast, the trend in product liability reform is to provide greater protection for business interests generally and for drug and device firms specifically. Probusiness interests rarely assert that injured individuals do not deserve compensation; rather, they argue that the liability prescription has adverse side effects and needs major alteration. They look for procedural and substantive changes in the system that will reduce
industry exposure to claims and the amount of awards available. On the other side, consumer groups and trial lawyers defend the present system. While no major liability reform has taken place in either the states or the federal government, limits on liability are often introduced. By the end of 1990, the reform environment looked inhospitable for device producers, with new regulation and no liability relief.
HR 3095, called the Safe Medical Devices Act, passed on 26 October 1990 and was signed into law by President Bush on November 17. Its original sponsors were Congressmen Henry A. Waxman and John D. Dingell, both powerful leaders in the field of product safety, health, and the environment.[15]
Henry Waxman is chair of the Subcommittee on Health and the Environment (part of the House Committee on Energy and Commerce). John Dingell is chair of the House Committee on Energy and Commerce and heads the Subcommittee on Oversight and Investigations.
When the bill was first introduced, it was accompanied by strong language challenging the credibility of both the device industry and the regulatory zeal of the agency. Representative Dingell described "the sorry inability of the FDA to implement the 1976 medical device law as Congress intended. The result has been that the health of the American public has been jeopardized. Indeed people—young babies, the elderly, and men and women in the prime of life—have died because unsafe medical devices have been allowed on the market without scrutiny."[16]
Congressional Record, 4 August 1989, E 2830.
Representative Waxman echoed this perspective: "It must come as an unhappy surprise to many to hear that, in 1989, we have no assurance that the medical devices used in, on, and around our bodies are safe."[17]Ibid., E 2815.
Why is the device industry on the defensive in Congress? There are many possible explanations. First, there have been several serious allegations against some manufacturers for careless manufacture and design, including defective infant monitors.[18]
See discussion in chapter 5.
However, none of the device problems has generated the kind of public concern that the generic drug industry scandals have in the same period.[19]In 1989, a scandal erupted in the generic drug industry when evidence that some firms had bribed FDA regulators with illegal gratuities. Two other firms admitted giving the FDA falsified data on their products. See Bruce Ingersoll and Gregory Stricharchuk, "Generic-Drug Scandal at the FDA Is Linked to Deregulation Drive," Wall Street Journal, 13 September 1989, 1, 10. See also Malcolm Gladwell, "A Few Bad Generic Drugs Can Spoil the Whole Industry," Washington Post National Weekly Edition, 21-27 August 1989, 31. See also American Enterprise Institute, Proposals for Reform of Drug Regulation Law, Legislative Analysis, no. 8 (Washington, D.C.: 1979).
It is ironic that consumer groups have generally supported these efforts until the AIDS crisis. AIDS activists have excoriated the FDA for being too slow in approving drugs for potentially lifesaving purposes, demanding less, not more, premarket scrutiny. No such forces are at work on the device side, so traditional congressional demands for greater safety have been unchallenged.
Furthermore, the device industry has not been very effective in counteracting congressional pressure, in part because of the fragmented nature of the industry—the diversity in firm size, in the focus on device products, and in the variations among the products themselves. This diversity often leads to a lack of a unified lobbying effort. Indeed, during the negotiations on the device legislation, some HIMA members broke away from the
organization. Sources reported that three HIMA members derailed negotiations with Dingell's oversight subcommittee, and counsel for the subcommittee alleged that the association had failed to represent the interests of all of its members. The subcommittee threatened to exclude HIMA from subsequent discussions on proposed legislation. HIMA claimed that it was trying to ensure consensus. However, the organizational infirmities within the association were quite evident.[20]
Healthweek, 10 November 1988, 7.
The principal purpose of the new law was to strengthen the Medical Device Amendments of 1976.[21]
House Committee on Energy and Commerce, Report to Accompany H.R. 3095, H. Rept. 101-808, 101st Cong., 2d sess. (5 October 1990). See also Senate Committee on Labor and Human Resources, Report to Accompany S. 3006, S. Rept. 101-513, 101st Cong., 2d sess. (9 October 1990).
The legislation contains many specific provisions, but all either increase FDA oversight and control of the device industry or streamline the detailed and often cumbersome provisions of the 1976 act. In the first category are provisions to increase the civil penalties that can be assessed against manufacturers for violations of the act, requirements for postmarket surveillance for permanent implants that pose serious risks or support human life, expanded reporting requirements, including the extension of mandatory reporting to "user facilities." These are defined to encompass hospitals, nursing homes, ambulatory surgical centers, and certain other outpatient facilities. The new law also makes it more difficult for a manufacturer to enter the market under "substantial equivalence" without the submission of safety and effectiveness data and codifies other current FDA practices.The more cumbersome Class II requirements have been streamlined, with rigid performance standard requirements giving way to more flexible "special controls." Performance standards are made discretionary, not mandatory, and the process is more efficient. The 1990 law addresses many details of FDA regulatory authority. Much was delegated to the agency. Exactly how the agency will implement the new law, how it will allocate resources, and whether it will emphasize enforcement or efficiency depend on many factors. At best, the medical device industry is left with additional regulatory uncertainty.
Reform of Product Liability
The trend toward greater regulation is in stark contrast to the politics of product liability reform. As we saw in chapter 6,
product liability rules derive from state court decisions and state legislative enactments. Product liability exposure can vary significantly among jurisdictions. However, the trend in virtually all the states is to limit the expansion of product liability that occurred in the 1970s. In addition, there are proposals to create a new federal product statute that would preempt state laws.
The press for reform in the 1990s derives from the perception of an insurance crisis that prevented producers from acquiring adequate and affordable coverage. The perceived crisis arose because of the massive expansion of liability standards, an explosive growth in the number of lawsuits, and increases in the size of jury awards in the 1970s.[22]
The Rand Corporation, Institute for Civil Justice, Tort Policy Working Report, February 1986.
In 1990, there was no consensus on the severity of either the liability or the insurance crisis.[23]Other studies have challenged these findings. The General Accounting Office issued a report in 1988 (Product Liability: Extent of Litigation Explosion in Federal Courts Questioned) that found that, at least in federal court filings, most claims related to asbestos (40 percent of growth), the Dalkon Shield (12 percent), and bendectin (5 percent).
Despite the disputes about the real impact of liability, business and insurance interests have kept up a steady pressure to reform the system, particularly in the area of product liability. There have been notable successes in state legislatures. As of 1990, thirty-nine states had passed some form of liability reform,[24]
Sheila R. Shulman, "Tort Reform Activities in the United States," in Sheila R. Shulman and Louis Lasagna, eds., Trends in Product Liability Law and No-Fault Compensation for Drug-Induced Injuries (Boston: Center for the Study of Drug Development, 1990), 46-49.
including more procedural hurdles for plaintiffs, limits on punitive damages, and narrower definitions of the concept of defect. All are designed to provide more protection from liability for corporate defendants.Reforms in some states related specifically to drugs and devices. The most directly relevant provision is known as the "government standards" defense. For example, New Jersey's tort reform statute provides that drugs, devices, food, and food additives that have received premarket approval or are licensed or regulated by the FDA shall not be subject to punitive damages unless the product manufacturer or producer knowingly withheld information or misrepresented the product during the approval process.[25]
Ibid., citing An Act Concerning Product Liability and Punitives Damages, State of New Jersey, chap. 197, laws of 1987, 47.
Another provision in the New Jersey law provides a presumption that any FDA warning is adequate, thus weighing the evidence in favor of a producer who has allegedly failed to adequately warn the user of the product's risks.[26]Presumptions in this context mean a rule of law in which the court will draw a particular inference from a particular fact unless and until the truth of such inference is disproved. This means that the presumption works in favor of the defendant, assuming that there has been adequate warning, unless the plaintiff proves otherwise.
Federal legislation to establish a uniform set of national rules for product liability legislation has been introduced in Congress every year for ten years. Two bills were pending in 1990. If passed, the legislation would preempt state law and require state courts to enforce the federal provisions instead.
Of course, the provisions of each particular piece of legislation vary. The Product Liability Reform Act (Senate bill 1400) serves as an example of federal efforts at product liability reform. Of particular relevance to the device industry is the government standards defense, similar to the New Jersey rule discussed above. Because of the experience of Dalkon Shield claimants, previous bills had included bars to government standards defense for contraceptive drugs and devices. Indeed, a bar to the defense had been proposed in, but was removed from, S. 1400. A number of bills are pending in 1991 to reform medical malpractice; at least one includes medical product reform as well.[27]
S. 1400, the Product Liability Reform Act, was introduced in the Senate in July 1989. Congressional hearings on the measure got underway in January 1990. Attempts to pass similar legislation go back ten years. There were four medical malpractice bills pending in Congress in the summer of 1991, including S. 1386, introduced by Senators Durenberger, Danforth, and McCain, which includes a government standards defense for drugs and medical devices.
Because there is considerable uncertainty about the future of reform at the federal level, predictions about the next decade are risky. However, the interest groups that will fight this battle in the 1990s are clearly defined. The device industry is allied with a broad coalition of manufacturers and insurers, all of whom exert a substantial influence at both the state and the federal levels. Consumer groups and trial lawyers, who are particularly well-organized and vociferous opponents to reform, are arrayed on the opposite side. One tactic has been to challenge state product liability reform in the courts. For example, opponents of reform have challenged the constitutionality of state imposed caps on damage awards, with mixed results. Review of the constitutionality of punitive damages was pending before the U.S. Supreme Court in 1990.[28]
For a discussion of the Supreme Court's recent decisions involving review of punitive damages in a variety of contexts, see Andrew Frey, "Do Punitives Fit the Crime?" National Law Journal, 9 October 1989,13-14.
Device producers will benefit if the government standards defense is widely adopted in states or imposed on all states through federal legislation. The government standards defense provides a link between two very different institutions—the FDA and the courts—and recognizes the interaction between these two divergent sources of public policy. However, either institution can change its requirements without the other adjusting its rules. If the FDA alters its requirements for premarket approval, the justification for the government standards defense may weaken. As of 1990, however, device producers have more regulatory hurdles, including risks of substantial civil penalties for failure to comply, without any corresponding liability relief. Coordination among policies has not been considered and appears to have been overlooked.
The problem of policy proliferation is that there seems to be little formal or consistent coordination among the diverse sources. Whether one supports or opposes the new proposals is based on a value judgment—do they, or do they not, produce sufficient levels of safety, without undue impacts on cost, access, or innovation. The solution will be political, and it will turn upon the power of interest groups to influence Congress.
Policy Reform at Distribution
The value underlying government payment programs is access to health care services. Ideally, cost-containment policies do not threaten the value of access but only keep out waste and excess. The impact on technology producers is mixed: government payment supports use and government cost containment inhibits it. There is the possibility of either promotion or inhibition as government policies try to balance the goal of access within a cost-contained system.
As we have seen, federal and state payers have become the gatekeepers for many categories of devices. The categories most affected by government policy include products used primarily by elderly Medicare beneficiaries, expensive capital equipment used in hospitals, and devices that indisputably raise costs.[29]
It is ironic that before the advent of the prospective payment system, Medicare was a boon to producers of products used primarily by beneficiaries. Now, with cost containment, a market dominated by Medicare can be the most difficult.
The larger the Medicare market share, the more dependent the producer is on decisions by HCFA. For producers with some Medicare market, HCFA's decision can influence, but not control, the other private third-party payers. If HCFA or its carriers refuse to cover a particular technology, the private payers may feel less compelled to do so for their policyholders. As we have seen in our case studies, some devices have been adversely affected by government reimbursement decisions. Of course, others such as pacemakers and IOLs have significantly benefited by government policies, although cost containment will probably affect all technologies to some degree.The politics of reform affecting device distribution are significantly different. In contrast to the more comprehensive legislative proposals for regulatory and/or liability reform, it is unlikely that there will be a major overhaul of public payment programs.[30]
Of course, predictions are always risky in the health care field. If the PSS completely breaks down, hospitals become bankrupt in large numbers or new public health crises erupt, the system could fail, and a new one could be imposed. Any new system would inevitably include some decision-making mechanism for technology adoption and diffusion, however. Cost controls are not likely to go away no matter what kind of public payment system is adopted.
It is also unlikely that any change would specificallybenefit medical technology producers. The pressure to contain costs is simply too strong.
Device producers have been disadvantaged by cost containment. However, many powerful private and public interests want to see medical costs controlled and reduced. While insurers and producers are on the same side of the debate over product liability reform, insurers who pay the bills lobby heavily for cost containment. Hospitals and employers are also feeling the crunch.
The pharmaceutical industry remains a lukewarm ally because it has not yet been significantly affected by federal government programs.[31]
The position of the pharmaceutical industry could change in the future given proposals to include drugs within the Medicare coverage.
The real allies of device firms in this context are consumers, who individually want access to all available alternatives, no matter how costly.[32]Surveys indicate considerable ambivalence among consumers. In general, they say that cost control is necessary, but do not personally want to see any limitations on access to all forms of high-tech care.
The medical device industry has not embraced consumer groups, no doubt because of suspicions stemming from their adversarial relationship in the product liability and the regulatory reform arenas. Alliances between the device industry and physicians vary, depending on whether the new device replaces doctors' services or otherwise affects a preferred practice pattern. In other words, the adversaries are very powerful, and the allies are only fair-weather friends. And unanimity among firms is difficult to achieve because the impact of payment policies varies depending on device characteristics.Periodically, there are calls for reform within the DRG system. Suggestions include the creation of technology-specific DRGs and partial or temporary exemptions from DRGs for new technologies (reimbursement on the old cost-plus system for short periods of time, for example). Advocates, however, often admit that there is considerable risk that the cost-control mechanisms will be defeated by these types of exceptions.
For the most part, the political stance of device producers has been to manage within the system rather than to seek to overhaul it. Recall the efforts of Acuson to craft flexible locations and uses for their products. These efforts are case by case, as manufacturers try to persuade the HCFA to cover their products and place them in profitable DRGs. Recently, the strategy has been to generate information on the costs and the benefits of the new technology for government. The industry has given up opposition to data gathering through technology assessment. Instead,
it has been willing, albeit reluctantly in some cases, to participate in technology assessment programs. In other words, the goal is to prove how beneficial (and cost effective) the new product is rather than to defeat cost containment. Manufacturers have been increasingly willing to take their case to the public, hoping that public pressure for desirable technologies can be exerted against government gatekeepers at the HCFA.
Given the realities of cost containment, producers of cost-increasing technologies will continue to face many inhibitory pressures on product adoption and diffusion. There is virtually no likelihood that comprehensive reform efforts could restore the price insensitive, technology consuming environment of the late 1960s and early 1970s. However, the present HCFA coverage and pricing policies are seriously flawed. We have seen many examples of both overdiffusion and underdiffusion because of HCFA decisions. HCFA needs more flexibility so that it can experiment with coverage alternatives, and it needs more information about the costs of new technologies. Coverage and payment reforms are necessary and should be forthcoming in the 1990s as Congress begins to focus on the medical arms race.
Understanding Interactions
There is no reason to believe that multiple policy interventions per se are necessarily bad for the industry or those the industry serves. However, our brown-bag review considers possible adverse interactions among the various policies, and interactions could be improved at several points. Figure 31 identifies the areas of interaction discussed in this section.
One interaction occurs when two policies create overlapping, duplicate, or conflicting incentives (see A on figure 31). Both regulation and product liability are premised on the value of safety, yet their many differences—institutional, procedural, and methodological—lead to potential adverse reactions. Following a description of the interaction, a range of treatment options are presented.
Another form of interaction occurs when policies have similar goals but operate at different stages of innovation (see B on figure 31). That is, government regulation and government
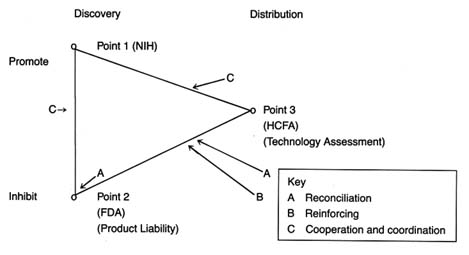
Figure 31. Policy interactions.
payment both may inhibit the flow of devices, but for different reasons. Reinforcing strategies can be developed to ensure that the policies do not operate at cross purposes but instead mutually support one another. Policies could be realigned to reinforce each other. The Pacemaker Registry, which is described below, is an example of reinforcing strategies, linking compliance with FDA regulation to favorable government payment and coverage decisions.
Finally, an interaction occurs when policies to promote devices confront policies to inhibit them (see C on figure 31). Examples include situations in which NIH promotion of innovation runs up against FDA requirements for safety and efficacy or in which NIH promotes devices for which HCFA may refuse to pay. Formal or informal mechanisms to ease the conflict or potential conflict at these points are possible. These efforts are characterized as coordination or cooperative strategies.
Reconciliation Strategies: Product Liability and Regulation
There is no fundamental reason why two or more prescriptions cannot serve the same goal. Controlling high cholesterol counts might require both dietary modification and a cholesterol lowering drug; treating coronary artery disease might require both
medication and surgical intervention. Similarly, in the policy arena, it is possible to have more than one policy serving the same goal without conflict.
Product liability and regulation both support the underlying value of product safety. Yet the prescriptions to accomplish that goal are very different on many dimensions. First, and quite important, the two policies define the standards of safety differently. Regulators at the FDA are concerned with public health; its staff is composed of scientists, aided by advisory panels of independent experts. The agency evaluates products prospectively by reviewing laboratory and clinical data developed by the manufacturers. The agency can monitor the products after marketing, requiring reporting of adverse reactions, and if a product later proves unsafe, it can be removed from the market. In essence, the FDA's safety determination is scientific and primarily prospective. The validity of its decisions depends upon accurate information presented both before and after marketing.
The product liability system determines safety quite differently. The evaluation is retrospective, after the harm has occurred and the plaintiff has sued. The injured individual presents his or her claim to a jury of laypersons, not scientists, although scientific experts can testify on certain points. Many of the findings depend upon legal, not scientific, determinations. For example, the legal concept of causation of harm is quite different from the scientific concept of causation.[33]
Ben F. Small, "Gaffing at a Thing Called Cause: Medico-Legal Conflicts in the Concept of Causation," Texas Law Review 31 (1953): 630-659, 655.
In most jurisdictions, compliance with FDA requirements for marketing is no defense against a legal determination of defect (that is, a product is unsafe).Second, the two systems overlap. Regulation tries to deter the production of unsafe products through premarket and postmarket controls. The FDA has substantial powers to impose civil and criminal penalties. Liability law also protects society through its intended deterrence function. Arguably, producers are deterred from producing defective products through fear of subsequent product liability suits. However, liability law also compensates individuals for harms incurred from unsafe products and has the capacity to punish producers as well.
The existence of the two overlapping systems produces contradictory incentives for industry. The social function of regulation
depends on information—gathering data before marketing and reporting problems once the product has been distributed. Without adequate and accurate information, the safety goals cannot be met. On the other hand, the threat of product liability suits intimidates producers. Even for manufacturers who are exonerated after a trial, the costs of defending a suit can be quite high. Thus, there are powerful incentives, even for responsible producers, to protect or to withhold information. And because compliance with FDA requirements is no defense to a product liability action, there are even fewer incentives to comply with the regulators.[34]
Compliance with FDA regulations would occur, however, if the government standards defense were widely imposed.
Finally, complying with regulation and defending against product liability actions (through insurance or costs of litigation and payments of awards) can be very expensive. As we noted in previous chapters, producing the required premarket approval data and complying with reporting and notification requirements can be very costly to a company. Of course, the decision to regulate concedes that the costs are worth it to achieve the desired level of safety. However, product liability is also expensive, and many of the costs associated with the system are procedural. Only a small percentage of the actual amount spent is transferred to the plaintiff if he or she prevails. Supporters of this system also maintain that the social costs of accidents should be covered by producers. The traditional justification is that producers can raise prices to cover the true social costs of their products.
In medical technology, however, policymakers cannot be cavalier about costs. Indeed, the costs of health care present a significant social issue in their own right. Medical devices are an integral part of the health care system. Costs associated with the public demand for safety must be evaluated against the equally pressing demand for widespread access to advanced medical technology and the undeniable cost pressures that all third-party payers face. The imposition of any unnecessary additional cost on a medical product is unacceptable. The imposition of too many barriers can deter innovation as well. This has already occurred in contraceptive and vaccine technology. Thus, we must be very confident that the systems support the fundamental value of safety and that they are worth the costs that they impose.[35]
Much of this discussion is relevant to policy reform for pharmaceuticals as well.
Can safety be protected while streamlining the two systems? Can costs of these policies be reduced? Several alternatives are possible.
One response is to support the government standards defense in product liability actions. This defense presumes that the FDA definition of safety is adequate. FDA critics are likely to oppose the substitution of its definition for that of the courts on the grounds that the FDA has failed in its mission. The benefit of a government standards defense is that it allows the systems to remain generally intact and does not require a massive overhaul of long-standing institutions. The problem, of course, is that keeping the systems intact preserves their other problems, including the facts that costs remain high (cases must be defended) and that information disincentives continue to exist.
Another alternative is to support more extensive national product liability reform to ensure uniformity at the federal level.
A more drastic solution would be a formal merging of regulation and liability into a single regulatory/compensatory system. A detailed proposal integrating the safety goals of both institutions has been presented.[36]
See Foote, "Product Liability." The proposal, in brief, is to expand FDA authority so that it can compensate for device related injuries and can refer cases of fraud to the courts to impose a punitive function. The producer would benefit from compliance with the FDA. Only those who fail to comply would face judicial remedies. Such a system could be extended to all medical technologies, including pharmaceuticals, and could also encompass medical malpractice concerns.
Such a plan is a variant of other compensation programs, such as state workers compensation plans that provide no-fault awards for work related injuries. Streamlining and controlling the impact of liability law on innovation characterizes the National Childhood Vaccine Program and forms the basis of recommendations to protect AIDS vaccine development from liability law that would deter innovation.[37]Aspects of the National Childhood Vaccine Injury Compensation Program are similar. See U.S. Congress, Hearings before the Senate Committee on Labor and Human Resources , 98th Cong., 1st sess., 1984, S. Rept. 98-1060. In that program, plaintiffs injured by certain vaccines can choose a traditional product liability remedy, with some limitations, or an expedited administrative proceeding. For an interesting proposal, see The Keystone Center, "Keystone AIDS Vaccine Liability Project Final Report" (Keystone, Colo.: Keystone Center, May 1990).
Supporters of compensation programs generally assert that there are serious flaws in the judicial system that hurt both plaintiffs and defendants. They go beyond a critique of the costs of the system, alleging that arbitrary factors such as geography, quality of lawyers, and wealth of defendants result in similarly situated plaintiffs receiving very different awards.[38]
See Danzon, Medical Malpractice. See also the work of Steven Sugarman critiquing the liability system, in particular, "Doing Away with Tort Law," California Law Review 73 (May 1985): 555-664.
They find only a capricious relationship between the amounts that plaintiffs recover and the seriousness of their injuries. In addition, there are long delays and risks of no recovery for injured people. Liability is expensive, and less than half of the premiums paid for liability insurance go to compensation.[39]The Interagency Task Force on Products Liability estimated that 40 percent of premiums go for underwriting expenses and profit and 20 percent of premiums go for loss-adjustment expenses; that leaves only forty cents of every premium available to compensate victims. See also Sugarman, "Doing Away," 591-600.
One major goal of a unified regulatory/compensatory system would be elimination of conflicting incentives for product producers.
The manufacturer would receive benefits for compliance with the FDA so that the flow of information on adverse reactions would be unhindered, providing greater safety for the consumer. Compensation could be made more uniform, predictable, and expeditious. Limits on awards would reduce the costs to producers overall and would encourage certainty and predictability without sacrificing the compensatory function of the law. Procedural efficiencies would reduce costs, allowing for lower product prices and reducing the risk of deterring innovation.
It would be naive to assume that such a system would be easy to construct or that there would be no opposition from powerful political forces. But if we recognize the inefficiencies and inequities of policy proliferation, we must remain receptive to alternative solutions.
Reinforcing Strategies: Regulation and Payment
There are situations in which two government policies inhibit technology for different purposes. Can these programs reinforce their diverse goals?
Congress recently enacted the Pacemaker Registry, which is an excellent example of the use of reinforcing strategies. The Pacemaker Reimbursement Review and Reform was an amendment to the Deficit Reduction Act of 1984.[40]
Public Law 98-369.
The goal was to obtain more information on pacemaker performance, and the law requires health care providers, as a condition of Medicare payment, to submit pacemaker information to an FDA registry. It permits the FDA to require that providers return explanted devices (devices removed from a patient after implantation) to the manufacturer and that manufacturers test returned devices and share the test results with providers.The purpose of the registry is to aid the federal payment program in determining when Medicare should pay for pacemaker implantation or explantation, assessing the performance of pacemakers and pacemaker leads, determining when manufacturers should inspect their pacemakers or leads, and performing studies on these devices. The data collected must include the following: manufacturer, model, and serial number of
the pacemaker; the date and location of the implantation or removal; any express or implied warranties for the pacemaker or leads; the patient who received the device; the physician who implanted or removed the device; and the hospital or other provider who is billing Medicare for the procedure. Reporting information to the registry will be required only if Medicare is requested to pay for the pacemaker, and the bill applies only to devices implanted or removed after the effective date of the implementing regulations. The law authorizes the FDA's Center for Devices and Radiological Health (CDRH) to require that FDA personnel be present while the manufacturer tests Medicare covered devices that may have failed.
The idea for this joint program developed as problems with pacemaker performance grew. The FDA funded a pacemaker registry at University of Southern California Medical Center from 1974 to 1981. The purpose was to gather experience data on pacemakers. Five medical centers participated, and the program included data on two hundred models of pulse generators produced by sixteen manufacturers. The original contract applied only to pacemaker pulse generators, but it was expanded in 1979 to include information about pacemaker leads. The data gathered by centers funded by the FDA supported the 1982 congressional investigations of fraud and abuse in the pacemaker industry.
The Center for Devices and Radiological Health set up a task force to determine how to proceed with the registry and to coordinate its efforts with HCFA. On 6 May 1986, HCFA and FDA proposed regulations for the registry.[41]
Cardiac Pacemaker Registry, 51 Federal Register 16792-16799 (6 May 1986).
Data in the registry could cover 170,000 to 200,000 devices each year. The FDA deferred development of regulations for certain discretionary sections of the act until the agency had some experience with the actual functioning of the registry.In 1987, HCFA and FDA published a final rule implementing the national registry.[42]
Final Rule, Cardiac Pacemaker Registry, 52 Federal Register 27756-27765 (23 July 1987).
Under the rule, physicians and providers of services must supply specified information for the pacemaker registry each time they implant, remove, or replace a pacemaker or a pacemaker lead in a Medicare patient. If the information is not submitted, the HCFA can deny payment. The FDA gets the data to monitor the long-term clinical performance of pacemakersand leads. The registry is used together with the more general medical device reporting (MDR) regulation to track failures or defects in certain models and to notify HCFA so that they may stop Medicare payments for those models.
There is no doubt that pacemakers provide important benefits to certain patients. Medicare pays for about 85,000–100,000 pacemaker implants and replacements every year, which account for a large percentage of Medicare costs. Pacemakers have also been linked to serious medical complications as well as to fraud and abuse in the payment system. The HCFA/FDA plan allows the efforts of each agency to reinforce the other's: linking payment to safety information both improves regulation and reduces fraud. Similar linkages ought to be considered for other high-risk, expensive, but highly beneficial, medical technologies.
Cooperation and Coordination: Promotion and Payment
Another policy interaction focuses on the relationship between programs that promote and programs that inhibit device flow. The NIH is the primary agency charged with promoting medical device discovery, and it has interacted with other federal agencies whose goals include inhibiting medical devices for a variety of reasons.
The NIH can interact with the FDA. If the NIH funded only investigator-initiated projects, there would be little opportunity for contact with the regulatory agency, which reviews the product only in its later stages of development. However, for some targeted programs, the NIH has become involved in designing clinical trials and supervising product testing activities. In these cases, the researcher may discover that the FDA has a strong interest in the design and progress of the trials. Occasionally turf battles erupt if the FDA disapproves of a trial design that has been developed in conjunction with the NIH.[43]
Sources have described such turf wars between the National Cancer Institute and the FDA. However, in the context of AIDS research, an emergency situation, the NCI and the FDA cooperated in designing research and approval mechanisms for new drugs.
Formal and informal communication is absolutely necessary to encourage technology transfer. But the perspectives of the two agencies are different: the NIH promotes and the FDA inhibits. With appropriate understanding, however, better cooperation could occur. There is evidence that such cooperation has developed in collaborative AIDS research. Perhaps because
of the perceived crisis, and pressure on the FDA to behave as a promoter rather than as an inhibitor of AIDS activists, most observers have been impressed by the efforts of both federal agencies to cooperate.
The NIH and HCFA also interact. Because these agencies operate at different ends of the innovation spectrum, the interaction is rarely direct. However, an issue has arisen regarding NIH promotion of complex technologies such as the artificial heart. In the short term, and probably in the long term as well, the artificial heart will remain an extraordinarily expensive technology. The question is whether federal dollars should be spent developing a product that the federal government may never be able to provide to all the elderly that might benefit from it. Supporters argue that many scientific discoveries can be linked to the AHP program. Still others recognize that the research would never take place without federal support. Should there be controls on the promotion of potentially cost-increasing technologies, those very products that HCFA is trying to discourage? Does that smack of industrial policy or of federal domination of research for cost-control, rather than scientific, purposes?
Should the NIH promote cost-reducing technologies? Even if the marketplace has incentives for such products, would federal support give greater weight to their development, particularly to devices that could significantly reduce costs? One technology that has been suggested for federal attention is the development of devices to treat incontinence, an area where treatment has not yet made significant progress but which poses a major and expensive problem for the elderly. Urinary incontinence is the involuntary loss of urine severe enough to have social and/or hygienic consequences. It is a significant cause of disability and dependence. The monetary costs of management are conservatively estimated at $10.3 billion annually. The estimates are that at least ten million adult Americans suffer from incontinence to some degree, including half of all nursing home patients.[44]
Biomedical Business International 12 (20 June 1989): 84.
Research is increasing the range of options, including medication, catheterization, urethral clamps, behavioral management, pelvic muscle exercises, implantation of an artificial urinary sphincter, electrotherapy, and surgery. Some therapies, such as collagen implants, cost over $1,800 for the kit, surely outside the
price range of incontinent patients admitted to a long-term care facility. It may be that the marketplace already encourages innovation for cost-reducing technologies. However, one could argue that the NIH could be enlisted in the cost-containment war as well.
This section supplies a framework for thinking about the policy environment. Policy proliferation can lead to problems; these problems can be addressed by efforts to reconcile, reinforce, and coordinate interactions. These efforts require an understanding of the different missions of the policy institutions and the broad political context in which they operate.
10
The Future
A good part of the tribulations of patients (and their physicians) comes from unreal attempts to transcend the possible; to deny its limits, and to seek the impossible: accommodation is more laborious and less exalted, and consists, in effect, of a painstaking exploration of the full range of the real and the possible.
Oliver Sacks Awakenings
We have examined the patient, evaluated the prescriptions, and considered modifications in the treatment.[1]
Opening quote from Oliver Sacks, Awakenings (New York: Harper Collins, 1990), 226.
Now is the time to reflect on the prognosis. Prognostication is always problematic, particularly so in the world of health. Wild cards can surprise even the most meticulous diagnostician. There can be unexpected increases in demand for services. A few years ago, it is unlikely that anyone could have predicted the impact of the AIDS epidemic on health care planning and services. Like Pearl Harbor, AIDS has precipitated a dramatic redefinition of priorities and reallocation of resources. Supply can change as well. The recent recognition that the United States has serious budgetary problems and the growing recession of the early 1990s have drastically increased pressures to limit health care expenditures.Uncertainty in the health care marketplace is exacerbated by unrealistic expectations. The compelling desire for good health and a long life has led to excessive demands upon medicine and medical technology. In the words of neurologist Oliver Sacks, medicine is asked to "transcend the possible." Obviously, there are limits to the power of machines. Despite what medical ethicist Daniel Callahan has referred to as our "touching faith in
technical fixes," medical devices will never meet all the expectations imposed upon them.[2]
Daniel Callahan, What Kind of Life: The Limits of Medical Progress (New York: Simon and Schuster, 1990), 101.
Without a realistic grasp of these limitations, we will be constantly disappointed. Unfortunately, dashed hopes create a tendency to blame devices for failing to live up to them. Just as medical technology cannot solve our battles with mortality, it cannot be held responsible for the failure to do so.
Similarly, the public needs to appreciate the dilemma facing innovators and manufacturers of medical products. These producers must respond to the economic realities of the private sector in which they operate. Manufacturers in a market economy try to expand their market share and make a profit. However, when a product is used for health care, the public traditionally has been uncomfortable with the consequences of the profit motive. Some have accused pharmaceutical companies of "unconscionable" profit. When the asking price exceeds what some customers (or some insurers) are willing or able to pay, is it unconscionable?
On the other hand, expansion of markets beyond what may be appropriate can have significant consequences for medical device consumers and the health care system as a whole. Adverse reactions to IUD implants in inappropriate candidates caused serious harm. In many respects, the traditional capitalist notions of markets can lead to socially irresponsible behavior. Supply-induced demand leads to waste and unnecessarily increases public and private expenditures.
Because of the value of these products and the expectations placed on their producers, public policy wrapped itself around the medical device industry. We must recall Lewis Mumford's wise words: "The machine itself makes no demands and holds out no promises; it is the human spirit that makes demands and keeps promises." Government policies and our public institutions must keep their promises. However, just as we have to learn to live with the limitations of technology, so we must understand the limitations of public policy in the United States.
Throughout this book, we have explored the dynamics of policy proliferation—the multiple, overlapping, often inconsistent interventions that reflect American history, political structure, and culture. Policy proliferation is inevitable in the American
system of government. As Richard Neustadt, an astute observer of politics, has observed, we have not so much a government of separated powers as "a government of separated institutions sharing power."[3]
Richard M. Neustadt, Presidential Power (New York: John Wiley, 1976), 101.
Shared power has led to multiple interventions in its exercise, at both the federal and the state levels of government.It is worth noting that shared power may offer benefits. For example, Justice Brandeis praised the states as "laboratories of experimentation." To some extent, our fragmented federal institutional structures may be laboratories as well. Experimentation implies distinctive solutions. Among these many alternatives, one of these laboratories of experimentation might get it right.
Tinkering with some unwanted effects of multiple public policies has been suggested in this book. Medical device innovators are tinkerers, and medical device, policy benefits from the tinkerer's art. As was discussed in chapter 9, incremental adjustments can greatly improve the policy environment. Incremental reform involves, in Sacks's words, laborious, painstaking accommodations, no less important because they are less dramatic than sweeping structural changes.
Tinkering with policy processes, however, should not be the end of the story because it does not address the hard questions that society has yet to face. Callahan's point is well taken: changing the mechanics of the system is no substitute for examination of "the psychological, moral, and political assumptions that lie below it."[4]
Callahan, What Kind of Life, 97.
In the final analysis, the most important issues are neither questions of science and technology nor simple questions of policy reform. The underlying issues reflect moral values about how we want to live and how we want to die, as individuals and as a society. We need on-going discussions of moral values that concern equity, resource allocation, and responsibility for adverse outcomes, to name just a few. The controversy surrounding the state of Oregon's attempt to prioritize Medicaid services arose from our difficulty in facing challenging moral issues.Medical device technology is integral to this moral debate. Indeed, consideration of the moral consequences has not kept pace with the ever-increasing capabilities of medical devices. Life-support equipment can keep human beings "alive" indefinitely,
but is this artificial extension of life compelled by moral or ethical principles? Does the existence of the machine require its use? Should economic resources be relevant to these considerations?
Before the advent of life-extending technologies, these decisions were left to God or fate. Now, in whose hands do these decisions reside? Are they exclusively individual decisions for families and physicians? The substantial role of public institutions has raised many of these individual dilemmas to the realm of public policy. Government payers must allocate limited resources. Who should participate in these policy decisions: physicians, theologians, politicians, judges, patients?
Many policy institutions are restricted by their narrow jurisdictions. Thus, for example, we debate minor alterations of regulation of particular devices and whether to lower reimbursement to producers of home medical equipment. We seem to quibble at the margins without facing these larger questions. It is imperative that we find appropriate forums for debate. The halls of the Food and Drug Administration or the Health Care Financing Administration are not the proper places to do so. Congress and state legislatures could begin the process, although it is important that the debates rise above purely partisan politics. Additional forums must be created where the people who will have to live with these important decisions can participate and help work toward a consensus on pivotal questions.
Medical devices are critical weapons in the armamentarium against disease. As a society, we want to encourage the flow of new, effective innovations. However, the challenge is to promote sufficient and useful medical weaponry without encouraging a medical arms race. Overuse and misuse, as we have seen, induce excess demand, waste valuable resources, and may harm the very people the weapons are designed to protect.
We must manage this medical arms race. This book has told the story of "adroit inventions and adaptations in politics" and their consequences to the products, the producers, and the public. The system is not perfect; considerable work remains to be done. Our expectations of both the devices and the policies that affect them, however, must recognize their limitations. Only then can the painstaking exploration of the real and the possible succeed.